README



Introduction
The main purpose of the PyPGx package is to provide a unified platform for pharmacogenomics (PGx) research. PyPGx is and always will be completely free and open source.
The package is written in Python, and supports both command line interface (CLI) and application programming interface (API) whose documentations are available at the Read the Docs.
Quick links:
PyPGx can predict PGx genotypes (e.g. *4/*5) and phenotypes (e.g.
Poor Metabolizer) using various genomic data, including data from
next-generation sequencing (NGS), single nucleotide polymorphism (SNP) array,
and long-read sequencing. Importantly, for NGS data the package can detect
structural variation (SV) using a machine learning-based
approach. Finally, note that PyPGx is compatible with both of the Genome
Reference Consortium Human (GRCh) builds, GRCh37 (hg19) and GRCh38 (hg38).
There are currently 88 pharmacogenes in PyPGx:
ABCB1 |
ABCG2 |
ACYP2 |
ADRA2A |
ADRB2 |
ANKK1 |
APOE |
ATM |
BCHE |
BDNF |
CACNA1S |
CFTR |
COMT |
CYP1A1 |
CYP1A2 |
CYP1B1 |
CYP2A6/CYP2A7 |
CYP2A13 |
CYP2B6/CYP2B7 |
CYP2C8 |
CYP2C9 |
CYP2C19 |
CYP2D6/CYP2D7 |
CYP2E1 |
CYP2F1 |
CYP2J2 |
CYP2R1 |
CYP2S1 |
CYP2W1 |
CYP3A4 |
CYP3A5 |
CYP3A7 |
CYP3A43 |
CYP4A11 |
CYP4A22 |
CYP4B1 |
CYP4F2 |
CYP17A1 |
CYP19A1 |
CYP26A1 |
DBH |
DPYD |
DRD2 |
F2 |
F5 |
G6PD |
GRIK1 |
GRIK4 |
GRIN2B |
GSTM1 |
GSTP1 |
GSTT1 |
HTR1A |
HTR2A |
IFNL3 |
IFNL3 |
ITGB3 |
ITPA |
MT-RNR1 |
MTHFR |
NAT1 |
NAT2 |
NUDT15 |
OPRK1 |
OPRM1 |
POR |
PTGIS |
RARG |
RYR1 |
SLC6A4 |
SLC15A2 |
SLC22A2 |
SLC28A3 |
SLC47A2 |
SLCO1B1 |
SLCO1B3 |
SLCO2B1 |
SULT1A1 |
TBXAS1 |
TPMT |
UGT1A1 |
UGT1A4 |
UGT1A6 |
UGT2B7 |
UGT2B15 |
UGT2B17 |
VKORC1 |
XPC |
Your contributions (e.g. feature ideas, pull requests) are most welcome.
Citation
If you use PyPGx in a published analysis, please report the program version and cite the following article:
Lee et al., 2022. ClinPharmSeq: A targeted sequencing panel for clinical pharmacogenetics implementation. PLOS ONE.
In this article, PyPGx was used to call star alleles for genomic DNA reference materials from the Centers for Disease Control and Prevention–based Genetic Testing Reference Materials Coordination Program (GeT-RM), where it showed almost 100% concordance with genotype results from previous works.
The development of PyPGx was heavily inspired by Stargazer, another star-allele calling tool developed by Steven when he was in his PhD program at the University of Washington. Therefore, please also cite the following articles:
Lee et al., 2019. Calling star alleles with Stargazer in 28 pharmacogenes with whole genome sequences. Clinical Pharmacology & Therapeutics.
Lee et al., 2018. Stargazer: a software tool for calling star alleles from next-generation sequencing data using CYP2D6 as a model. Genetics in Medicine.
Below is an incomplete list of publications which have used PyPGx:
Wroblewski et al., 2022. Pharmacogenetic variation in Neanderthals and Denisovans and implications for human health and response to medications. bioRxiv.
Botton et al., 2020. Phased Haplotype Resolution of the SLC6A4 Promoter Using Long-Read Single Molecule Real-Time (SMRT) Sequencing. Genes.
Support PyPGx
If you find my work useful, please consider becoming a sponsor.
Installation
Following packages are required to run PyPGx:
Package |
Anaconda |
PyPI |
|---|---|---|
|
✅ |
✅ |
|
✅ |
✅ |
|
✅ |
❌ |
There are various ways you can install PyPGx. The recommended way is via conda (Anaconda):
$ conda install -c bioconda pypgx
Above will automatically download and install all the dependencies as well.
Alternatively, you can use pip (PyPI) to install
PyPGx and all of its dependencies except openjdk (i.e. Java JDK must be
installed separately):
$ pip install pypgx
Finally, you can clone the GitHub repository and then install PyPGx locally:
$ git clone https://github.com/sbslee/pypgx
$ cd pypgx
$ pip install .
The nice thing about this approach is that you will have access to
development versions that are not available in Anaconda or PyPI. For example,
you can access a development branch with the git checkout command. When
you do this, please make sure your environment already has all the
dependencies installed.
Note
Beagle
is one of the default software tools used by PyPGx for haplotype phasing
SNVs and indels. The program is freely available and published under the
GNU General Public License. Users do not need to download Beagle separately
because a copy of the software (beagle.22Jul22.46e.jar) is already
included in PyPGx.
Warning
You’re not done yet! Keep scrolling down to obtain the resource bundle for PyPGx, which is essential for running the package.
Resource bundle
Starting with the 0.12.0 version, reference haplotype panel files and
structural variant classifier files in PyPGx are moved to the
pypgx-bundle repository
(only those files are moved; other files such as allele-table.csv and
variant-table.csv are intact). Therefore, the user must clone the
pypgx-bundle repository with matching PyPGx version to their home
directory in order for PyPGx to correctly access the moved files (i.e. replace
x.x.x with the version number of PyPGx you’re using, such as 0.18.0):
$ cd ~
$ git clone --branch x.x.x --depth 1 https://github.com/sbslee/pypgx-bundle
This is undoubtedly annoying, but absolutely necessary for portability reasons because PyPGx has been growing exponentially in file size due to the increasing number of genes supported and their variation complexity, to the point where it now exceeds upload size limit for PyPI (100 Mb). After removal of those files, the size of PyPGx has reduced from >100 Mb to <1 Mb.
Starting with version 0.22.0, you can now specify a custom location for the
pypgx-bundle directory instead of using the home directory. This can be
achieved by setting the bundle location using the PYPGX_BUNDLE environment
variable:
$ export PYPGX_BUNDLE=/path/to/pypgx-bundle
Structural variation detection
Many pharmacogenes are known to have structural variation (SV) such as gene deletions, duplications, and hybrids. You can visit the Genes page to see the list of genes with SV.
Some of the SV events can be quite challenging to detect accurately with NGS data due to misalignment of sequence reads caused by sequence homology with other gene family members (e.g. CYP2D6 and CYP2D7). PyPGx attempts to address this issue by training a support vector machine (SVM)-based multiclass classifier using the one-vs-rest strategy for each gene for each GRCh build. Each classifier is trained using copy number profiles of real NGS samples as well as simulated ones, including those from 1KGP and GeT-RM.
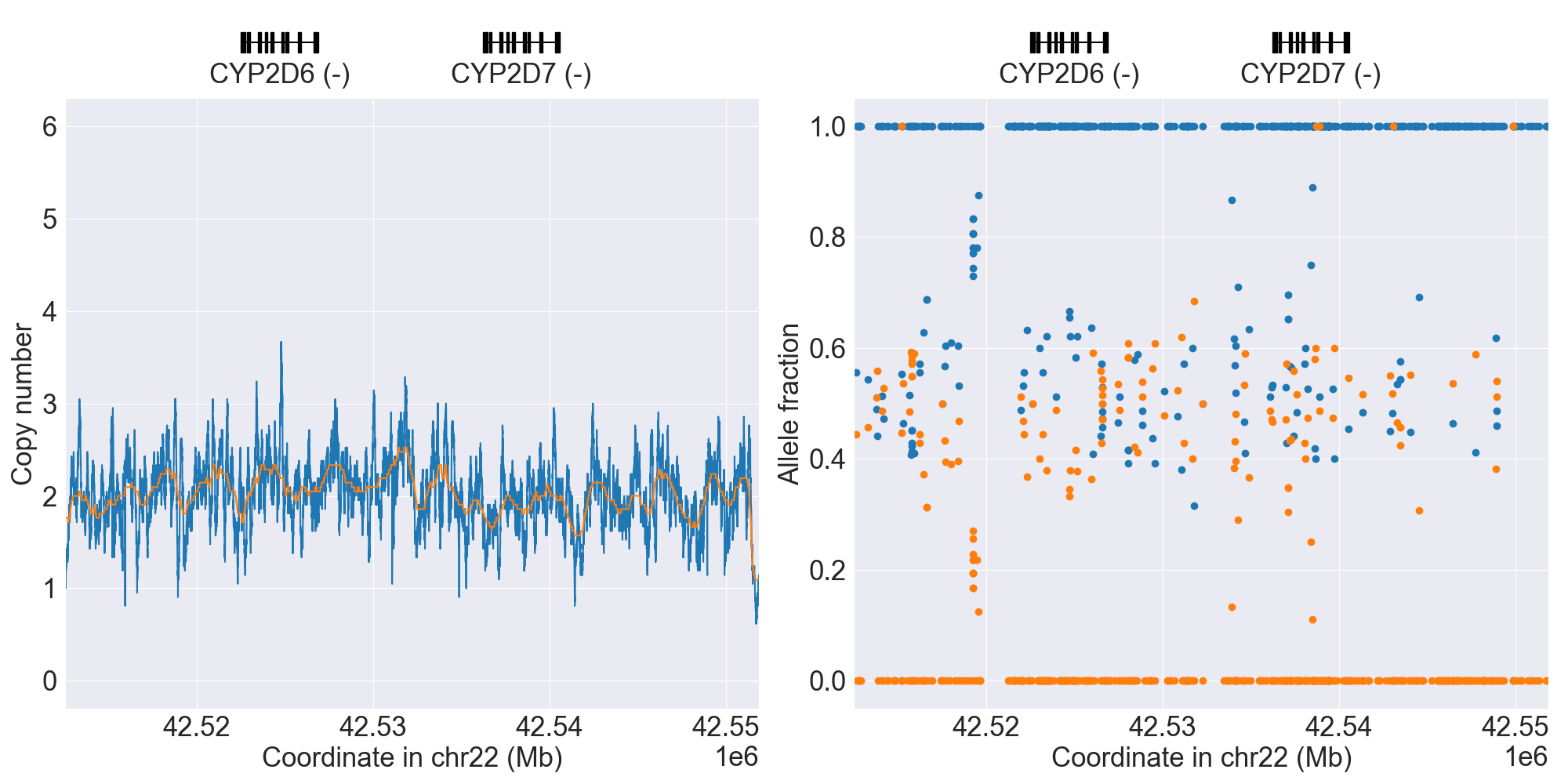
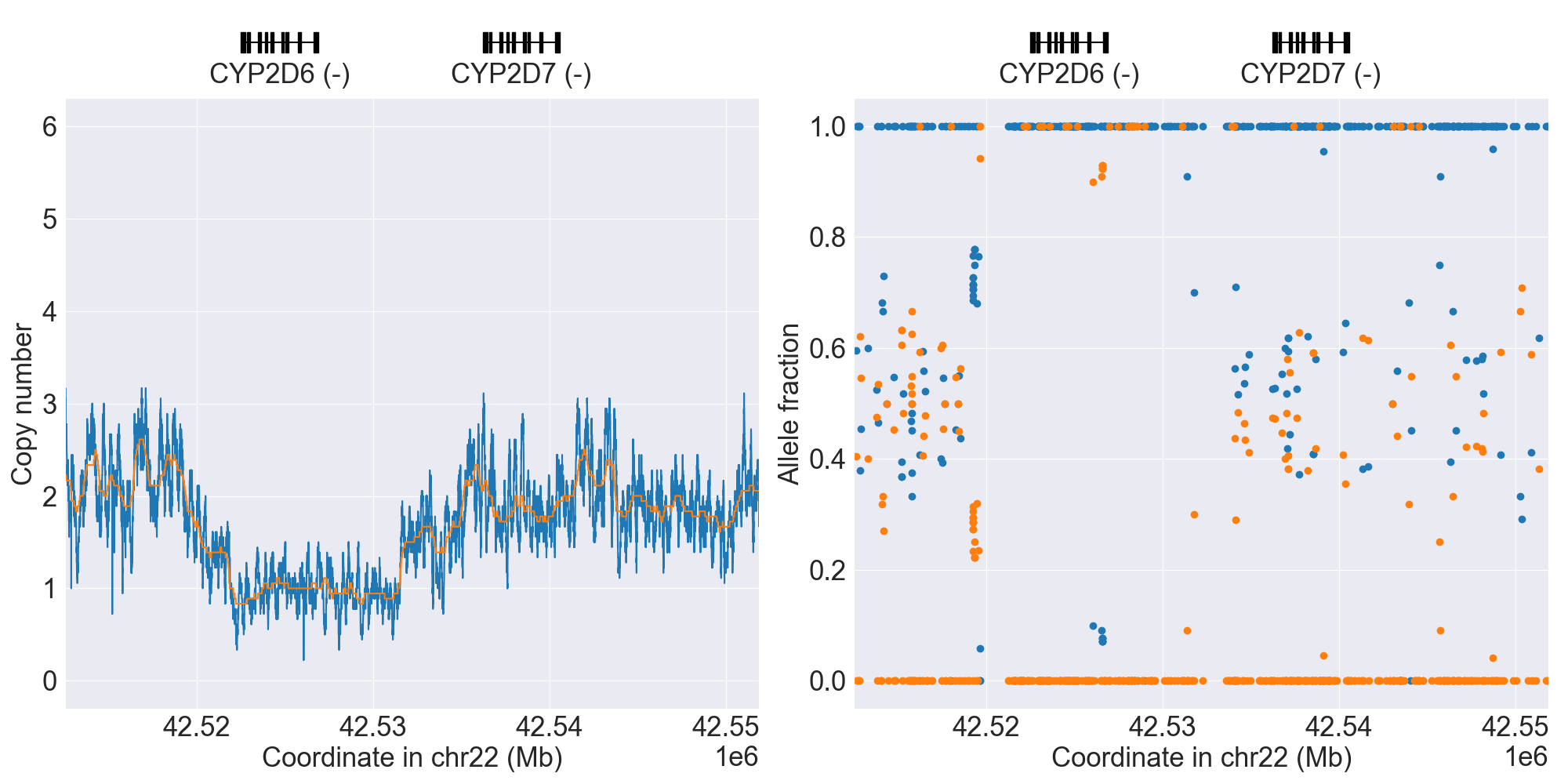
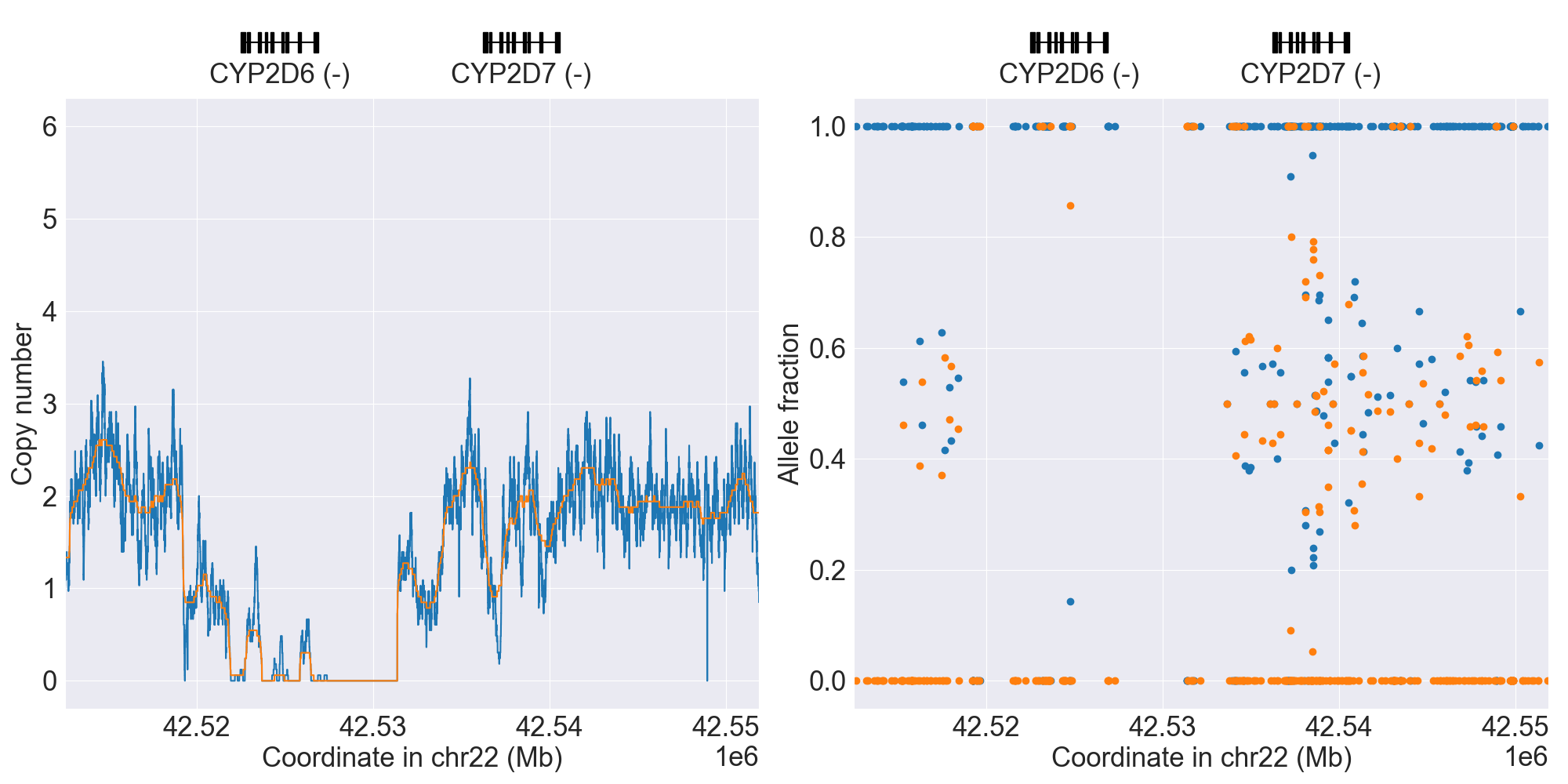
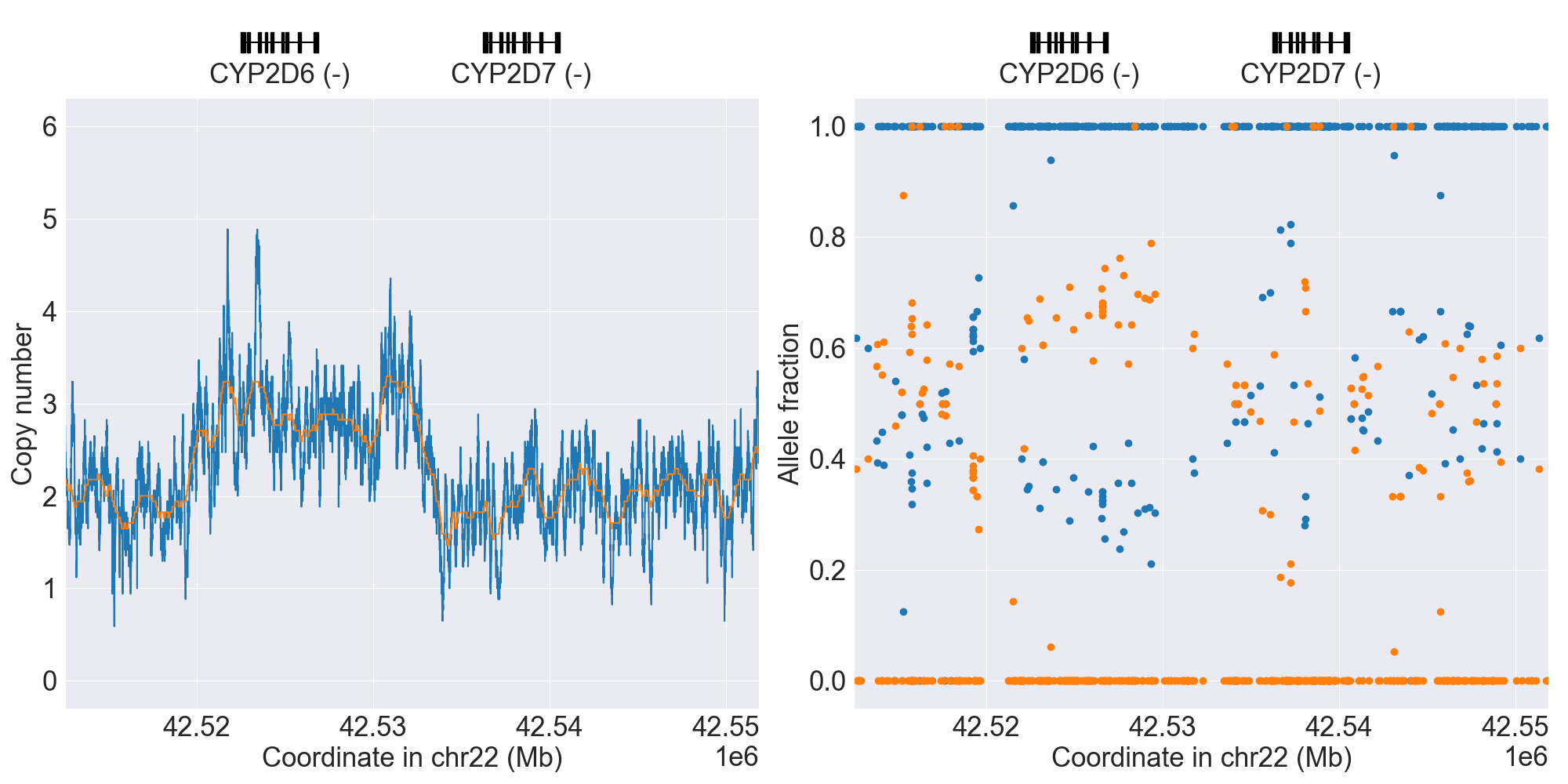
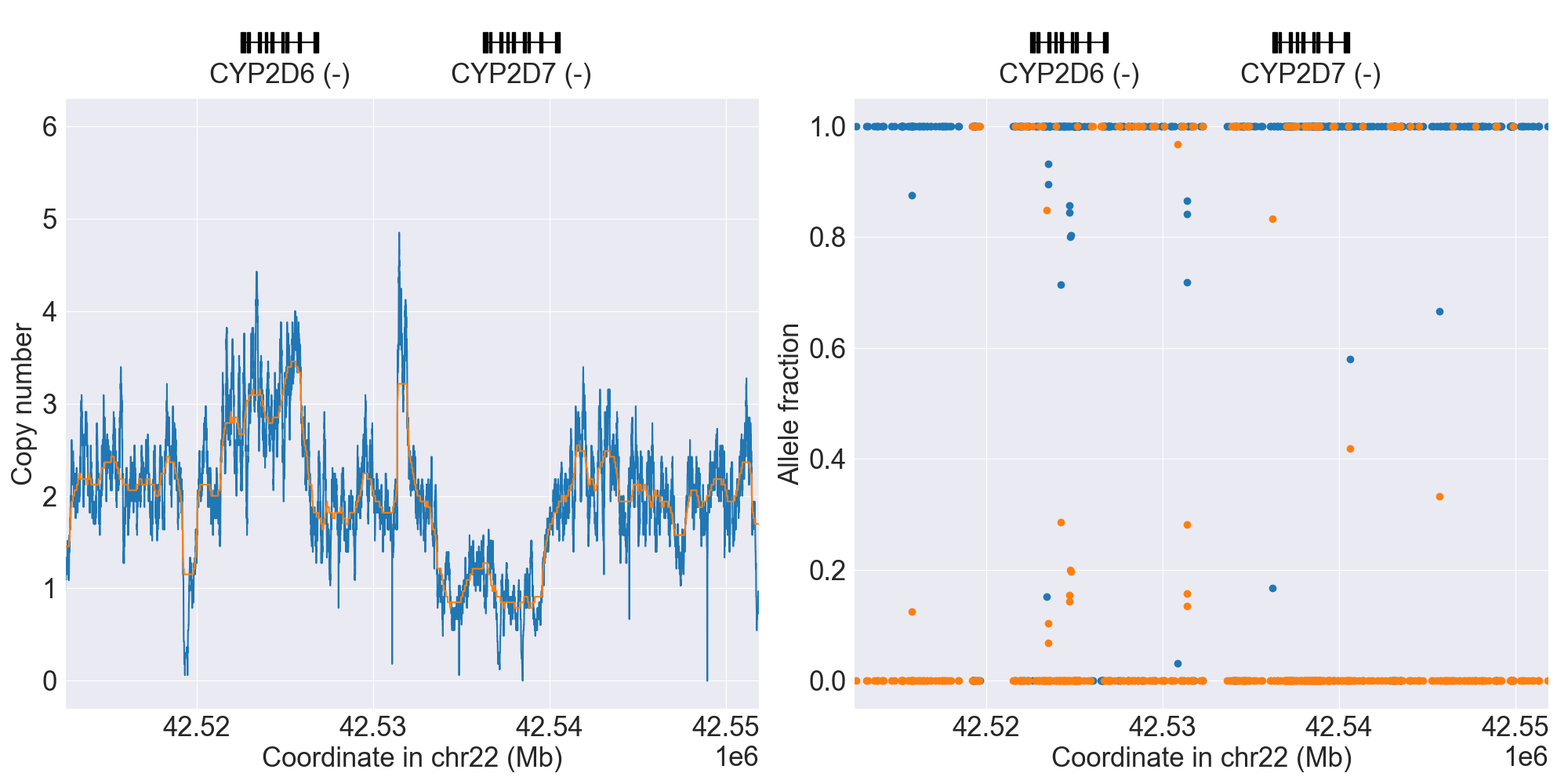
You can plot copy number profile and allele fraction profile with PyPGx to visually inspect SV calls. Below are CYP2D6 examples:
SV Name |
Gene Model |
Profile |
|---|---|---|
Normal |
|
|
WholeDel1 |

|
|
WholeDel1Hom |

|
|
WholeDup1 |
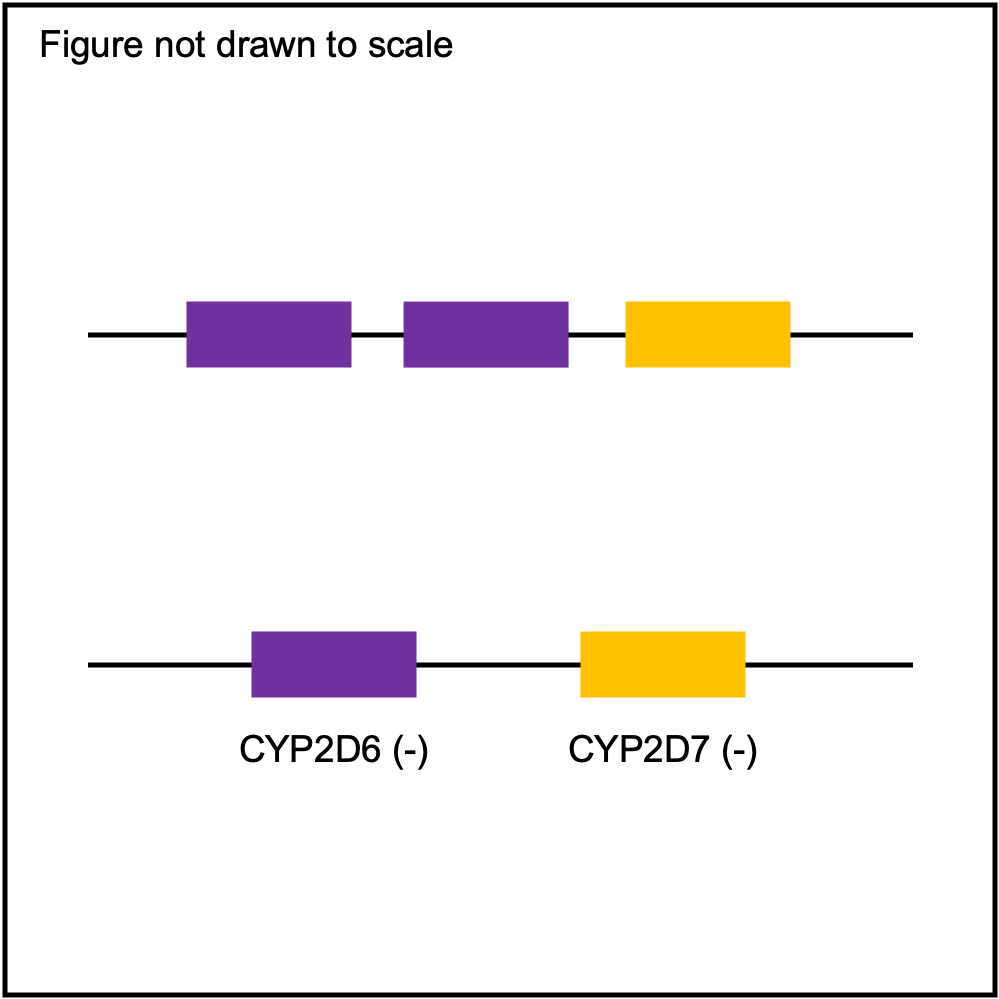
|
|
Tandem3 |
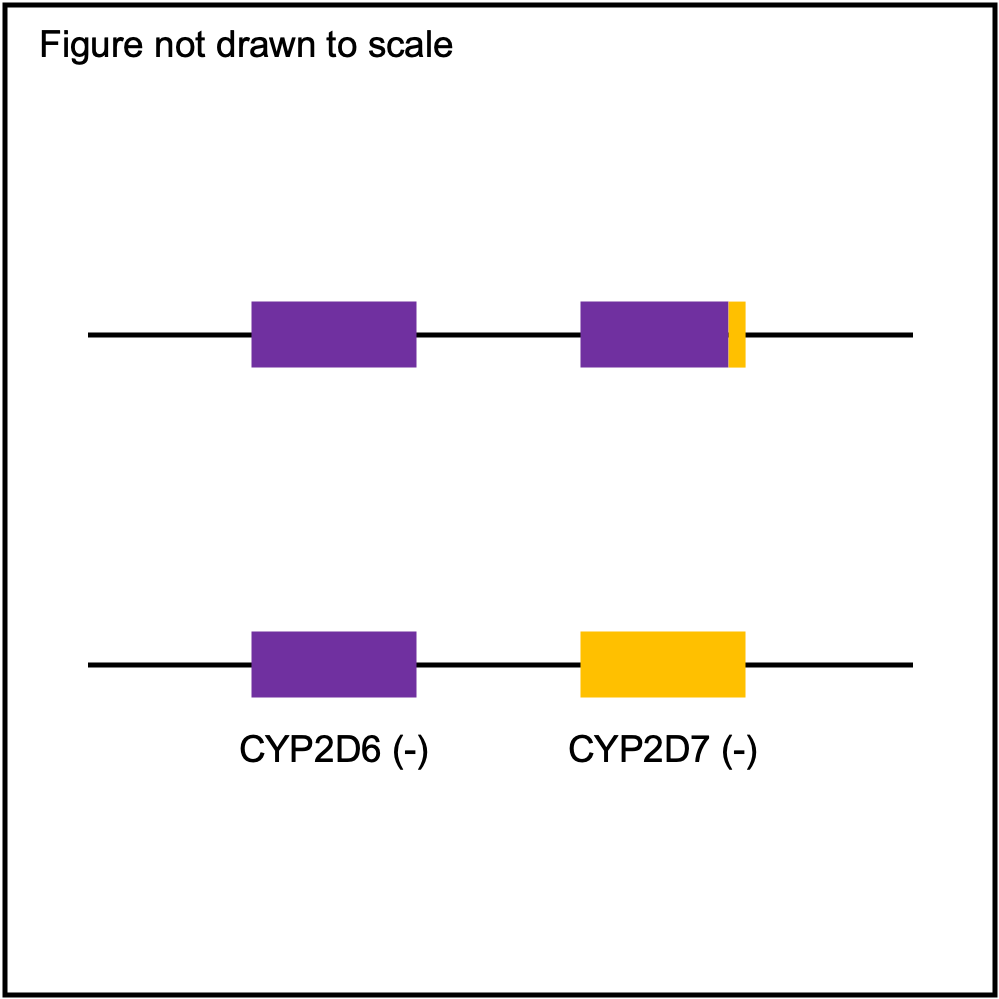
|
|
Tandem2C |
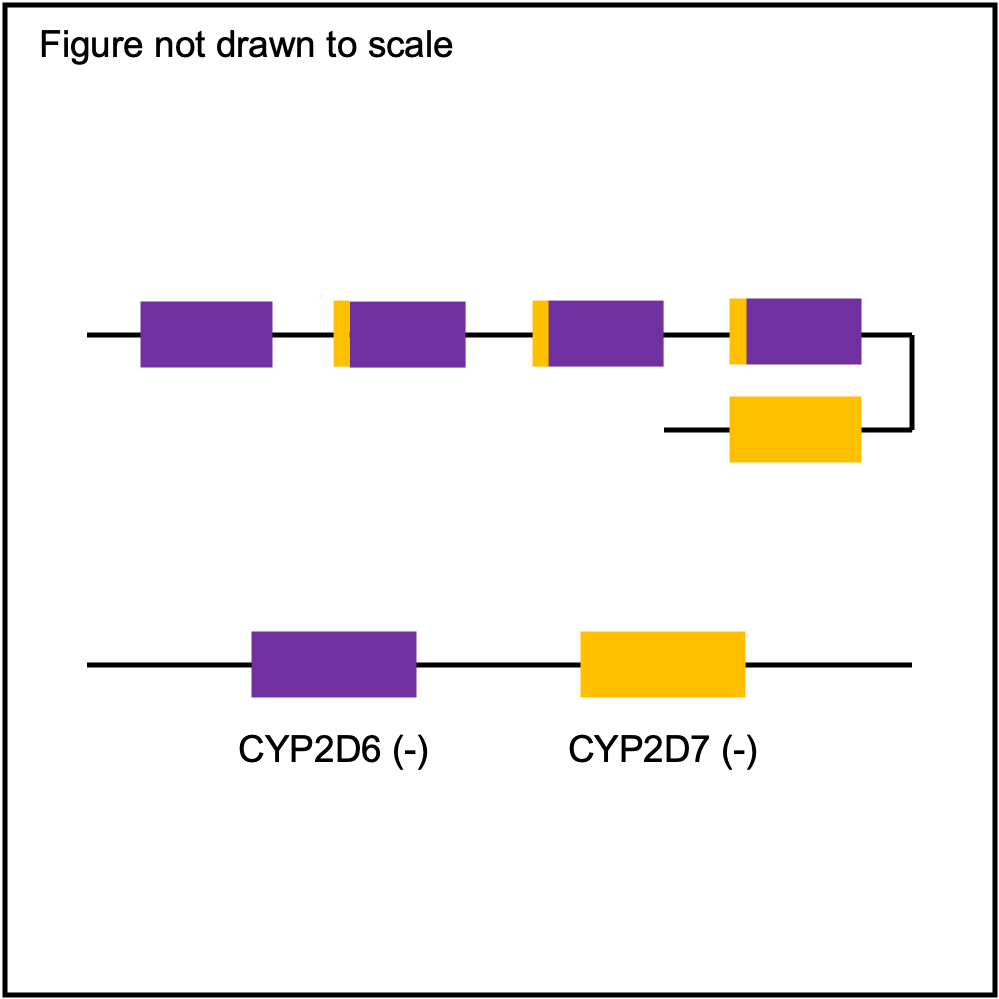
|
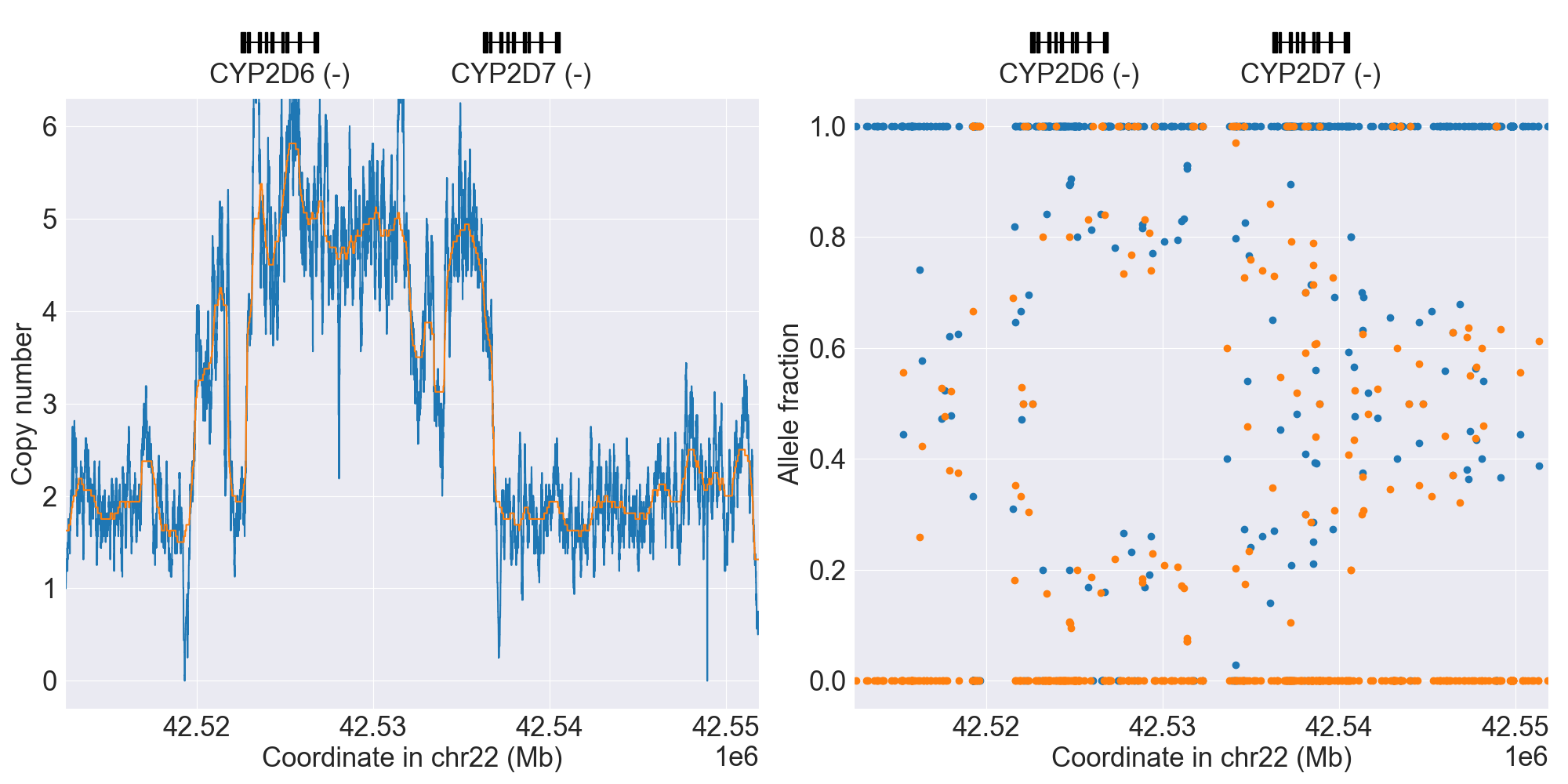
|
PyPGx was recently applied to the entire high-coverage WGS dataset from 1KGP (N=2,504). Click here to see individual SV calls, and corresponding copy number profiles and allele fraction profiles.
GRCh37 vs. GRCh38
When working with PGx data, it’s not uncommon to encounter a situation
where you are handling GRCh37 data in one project but GRCh38 in another. You
may be tempted to use tools like LiftOver to convert GRCh37 to GRCh38, or
vice versa, but deep down you know it’s going to be a mess (and please don’t
do this). The good news is, PyPGx supports both of the builds!
In many PyPGx actions, you can simply indicate which genome build to use. For
example, for GRCh38 data you can use --assembly GRCh38 in CLI and
assembly='GRCh38' in API. Note that GRCh37 will always be the
default. Below is an example of using the API:
>>> import pypgx
>>> pypgx.list_variants('CYP2D6', alleles=['*4'], assembly='GRCh37')
['22-42524947-C-T']
>>> pypgx.list_variants('CYP2D6', alleles=['*4'], assembly='GRCh38')
['22-42128945-C-T']
However, there is one important caveat to consider if your sequencing data is
GRCh38. That is, sequence reads must be aligned only to the main contigs
(i.e. chr1, chr2, …, chrX, chrY), and not to the
alternative (ALT) contigs such as chr1_KI270762v1_alt. This is because
the presence of ALT contigs reduces the sensitivity of variant calling
and many other analyses including SV detection. Therefore, if you have
sequencing data in GRCh38, make sure it’s aligned to the main contigs only.
The only exception to above rule is the GSTT1 gene, which is located on
chr22 for GRCh37 but on chr22_KI270879v1_alt for GRCh38. This gene is
known to have an extremely high rate of gene deletion polymorphism in the
population and thus requires SV analysis. Therefore, if you are interested in
genotyping this gene with GRCh38 data, then you must include that contig
when performing read alignment. To this end, you can easily filter your
reference FASTA file before read alignment so that it only contains the main
contigs plus the ALT contig. If you don’t know how to do this, here’s one way
using the fuc program (which should have already been installed along
with PyPGx):
$ cat contigs.list
chr1
chr2
...
chrX
chrY
chr22_KI270879v1_alt
$ fuc fa-filter in.fa --contigs contigs.list > out.fa
Archive file, semantic type, and metadata
In order to efficiently store and transfer data, PyPGx uses the ZIP archive
file format (.zip) which supports lossless data compression. Each archive
file created by PyPGx has a metadata file (metadata.txt) and a data file
(e.g. data.tsv, data.vcf). A metadata file contains important
information about the data file within the same archive, which is expressed
as pairs of =-separated keys and values (e.g. Assembly=GRCh37):
Metadata |
Description |
Examples |
|---|---|---|
|
Reference genome assembly. |
|
|
Control gene. |
|
|
Target gene. |
|
|
Genotyping platform. |
|
|
Name of the phasing program. |
|
|
Samples used for inter-sample normalization. |
|
|
Semantic type of the archive. |
|
Semantic types
Notably, all archive files have defined semantic types, which allows us to ensure that the data that is passed to a PyPGx command (CLI) or method (API) is meaningful for the operation that will be performed. Below is a list of currently defined semantic types:
CovFrame[CopyNumber]CovFrame for storing target gene’s per-base copy number which is computed from read depth with control statistics.
Requires following metadata:
Gene,Assembly,SemanticType,Platform,Control,Samples.
CovFrame[DepthOfCoverage]CovFrame for storing read depth for all target genes with SV.
Requires following metadata:
Assembly,SemanticType,Platform.
CovFrame[ReadDepth]CovFrame for storing read depth for single target gene.
Requires following metadata:
Gene,Assembly,SemanticType,Platform.
Model[CNV]Model for calling CNV in target gene.
Requires following metadata:
Gene,Assembly,SemanticType,Control.
SampleTable[Alleles]TSV file for storing target gene’s candidate star alleles for each sample.
Requires following metadata:
Platform,Gene,Assembly,SemanticType,Program.
SampleTable[CNVCalls]TSV file for storing target gene’s CNV call for each sample.
Requires following metadata:
Gene,Assembly,SemanticType,Control.
SampleTable[Genotypes]TSV file for storing target gene’s genotype call for each sample.
Requires following metadata:
Gene,Assembly,SemanticType.
SampleTable[Phenotypes]TSV file for storing target gene’s phenotype call for each sample.
Requires following metadata:
Gene,SemanticType.
SampleTable[Results]TSV file for storing various results for each sample.
Requires following metadata:
Gene,Assembly,SemanticType.
SampleTable[Statistics]TSV file for storing control gene’s various statistics on read depth for each sample. Used for converting target gene’s read depth to copy number.
Requires following metadata:
Control,Assembly,SemanticType,Platform.
VcfFrame[Consolidated]VcfFrame for storing target gene’s consolidated variant data.
Requires following metadata:
Platform,Gene,Assembly,SemanticType,Program.
VcfFrame[Imported]VcfFrame for storing target gene’s raw variant data.
Requires following metadata:
Platform,Gene,Assembly,SemanticType.
VcfFrame[Phased]VcfFrame for storing target gene’s phased variant data.
Requires following metadata:
Platform,Gene,Assembly,SemanticType,Program.
Working with archive files
To demonstrate how easy it is to work with PyPGx archive files, below we will
show some examples. First, download an archive to play with, which has
SampleTable[Results] as semantic type:
$ wget https://raw.githubusercontent.com/sbslee/pypgx-data/main/getrm-wgs-tutorial/grch37-CYP2D6-results.zip
Let’s print its metadata:
$ pypgx print-metadata grch37-CYP2D6-results.zip
Gene=CYP2D6
Assembly=GRCh37
SemanticType=SampleTable[Results]
Now print its main data (but display first sample only):
$ pypgx print-data grch37-CYP2D6-results.zip | head -n 2
Genotype Phenotype Haplotype1 Haplotype2 AlternativePhase VariantData CNV
HG00276_PyPGx *4/*5 Poor Metabolizer *4;*10;*74;*2; *10;*74;*2; ; *4:22-42524947-C-T:0.913;*10:22-42526694-G-A,22-42523943-A-G:1.0,1.0;*74:22-42525821-G-T:1.0;*2:default; DeletionHet
We can unzip it to extract files inside (note that tmpcty4c_cr is the
original folder name):
$ unzip grch37-CYP2D6-results.zip
Archive: grch37-CYP2D6-results.zip
inflating: tmpcty4c_cr/metadata.txt
inflating: tmpcty4c_cr/data.tsv
We can now directly interact with the files:
$ cat tmpcty4c_cr/metadata.txt
Gene=CYP2D6
Assembly=GRCh37
SemanticType=SampleTable[Results]
$ head -n 2 tmpcty4c_cr/data.tsv
Genotype Phenotype Haplotype1 Haplotype2 AlternativePhase VariantData CNV
HG00276_PyPGx *4/*5 Poor Metabolizer *4;*10;*74;*2; *10;*74;*2; ; *4:22-42524947-C-T:0.913;*10:22-42526694-G-A,22-42523943-A-G:1.0,1.0;*74:22-42525821-G-T:1.0;*2:default; DeletionHet
We can easily create a new archive:
$ zip -r grch37-CYP2D6-results-new.zip tmpcty4c_cr
adding: tmpcty4c_cr/ (stored 0%)
adding: tmpcty4c_cr/metadata.txt (stored 0%)
adding: tmpcty4c_cr/data.tsv (deflated 84%)
$ pypgx print-metadata grch37-CYP2D6-results-new.zip
Gene=CYP2D6
Assembly=GRCh37
SemanticType=SampleTable[Results]
Phenotype prediction
Many genes in PyPGx have a genotype-phenotype table available from the Clinical Pharmacogenetics Implementation Consortium (CPIC) or the Pharmacogenomics Knowledge Base (PharmGKB). PyPGx uses these tables to perform phenotype prediction with one of the two methods:
Method 1. Simple diplotype-phenotype mapping: This method directly uses the diplotype-phenotype mapping as defined by CPIC or PharmGKB. Using the CYP2B6 gene as an example, the diplotypes *6/*6, *1/*29, *1/*2, *1/*4, and *4/*4 correspond to Poor Metabolizer, Intermediate Metabolizer, Normal Metabolizer, Rapid Metabolizer, and Ultrarapid Metabolizer.
Method 2. Summation of haplotype activity scores: This method uses a standard unit of enzyme activity known as an activity score. Using the CYP2D6 gene as an example, the fully functional reference *1 allele is assigned a value of 1, decreased-function alleles such as *9 and *17 receive a value of 0.5, and nonfunctional alleles including *4 and *5 have a value of 0. The sum of values assigned to both alleles constitutes the activity score of a diplotype. Consequently, subjects with *1/*1, *1/*4, and *4/*5 diplotypes have an activity score of 2 (Normal Metabolizer), 1 (Intermediate Metabolizer), and 0 (Poor Metabolizer), respectively.
Please visit the Genes page to see the list of genes with a genotype-phenotype table and each of their prediction method.
To perform phenotype prediction with the API, you can use the
pypgx.predict_phenotype method:
>>> import pypgx
>>> pypgx.predict_phenotype('CYP2D6', '*4', '*5') # Both alleles have no function
'Poor Metabolizer'
>>> pypgx.predict_phenotype('CYP2D6', '*5', '*4') # The order of alleles does not matter
'Poor Metabolizer'
>>> pypgx.predict_phenotype('CYP2D6', '*1', '*22') # *22 has uncertain function
'Indeterminate'
>>> pypgx.predict_phenotype('CYP2D6', '*1', '*1x2') # Gene duplication
'Ultrarapid Metabolizer'
To perform phenotype prediction with the CLI, you can use the
call-phenotypes command. It takes a SampleTable[Genotypes] file as
input and outputs a SampleTable[Phenotypes] file:
$ pypgx call-phenotypes genotypes.zip phenotypes.zip
Pipelines
PyPGx currently provides three pipelines for performing PGx genotype analysis
of single gene for one or multiple samples: NGS pipeline, chip pipeline, and
long-read pipeline. In additional to genotyping, each pipeline will perform
phenotype prediction based on genotype results. All pipelines are compatible
with both GRCh37 and GRCh38 (e.g. for GRCh38 use --assembly GRCh38 in CLI
and assembly='GRCh38' in API).
NGS pipeline
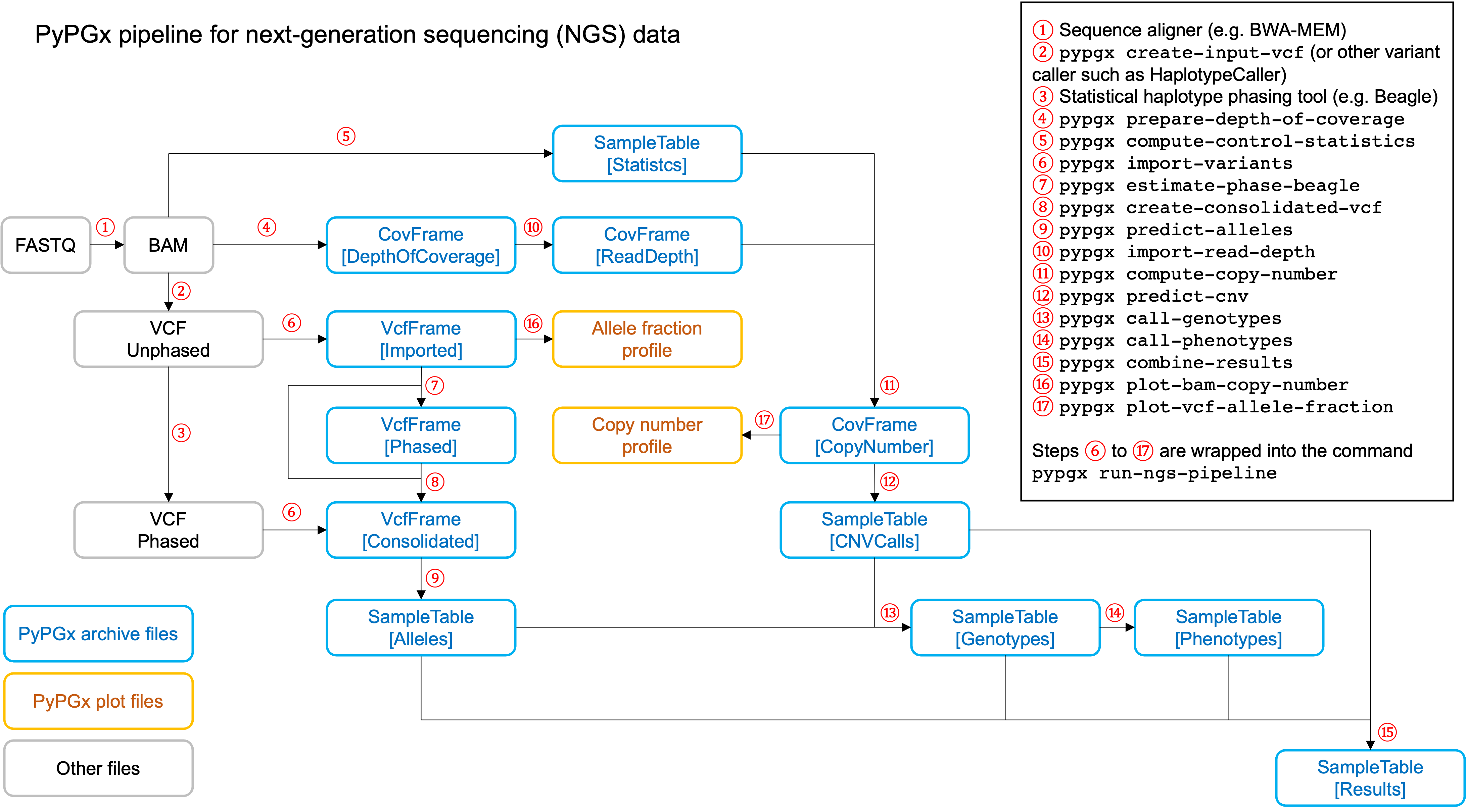
Implemented as pypgx run-ngs-pipeline in CLI and
pypgx.pipeline.run_ngs_pipeline in API, this pipeline is designed for
processing short-read data (e.g. Illumina). Users must specify whether the
input data is from whole genome sequencing (WGS) or targeted sequencing
(custom targeted panel sequencing or whole exome sequencing).
This pipeline supports SV detection based on copy number analysis for genes
that are known to have SV. Therefore, if the target gene is associated with
SV (e.g. CYP2D6) it’s strongly recommended to provide a
CovFrame[DepthOfCoverage] file and a SampleTable[Statistics] file in
addtion to a VCF file containing SNVs/indels. If the target gene is not
associated with SV (e.g. CYP3A5) providing a VCF file alone is enough. You can
visit the Genes page
to see the full list of genes with SV. For details on SV detection algorithm,
please see the Structural variation detection section.
When creating a VCF file (containing SNVs/indels) from BAM files, users have
a choice to either use the pypgx create-input-vcf command (strongly
recommended) or a variant caller of their choice (e.g. GATK4
HaplotypeCaller). See the Variant caller choice section for detailed
discussion on when to use either option.
Check out the GeT-RM WGS tutorial to see this pipeline in action.
Chip pipeline
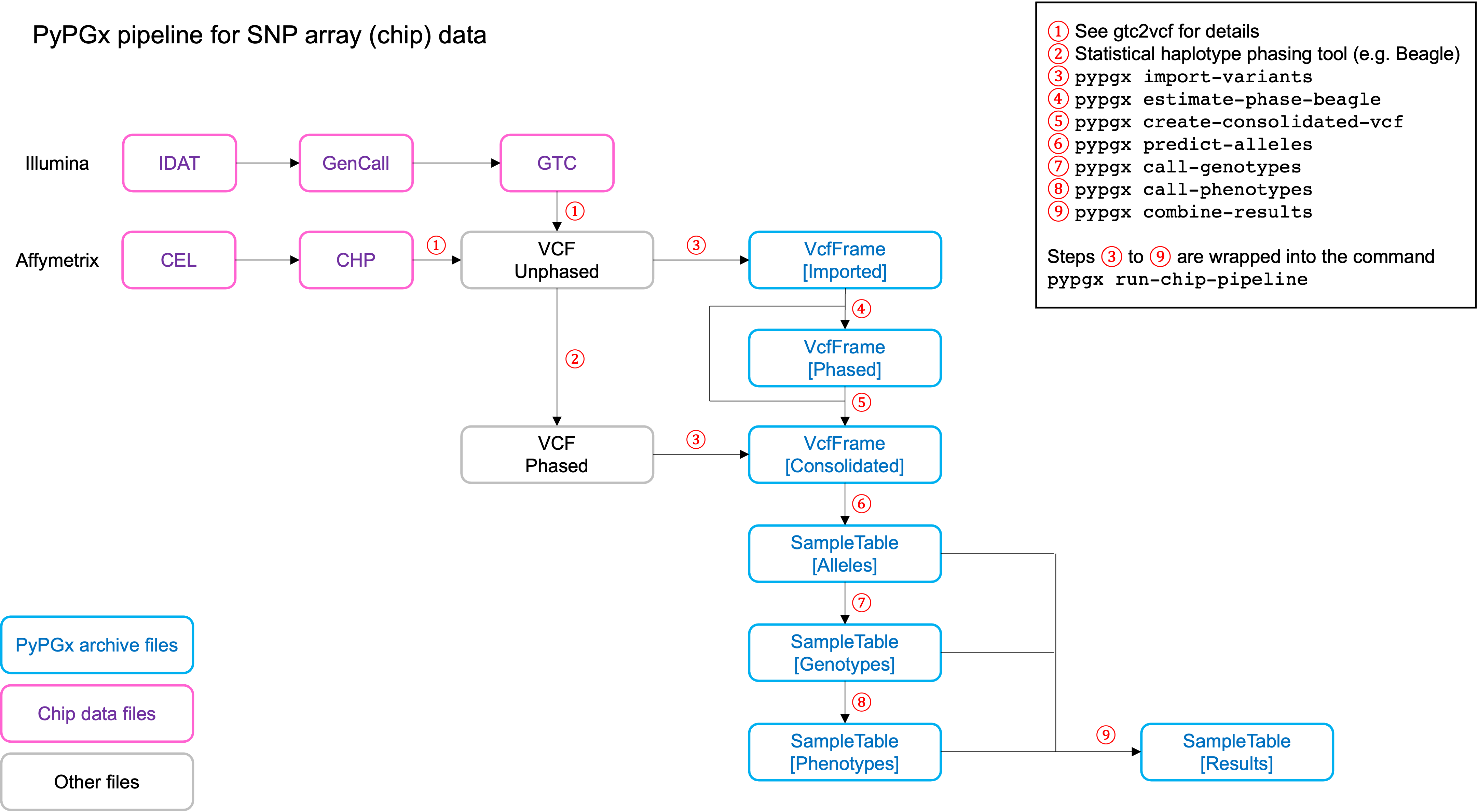
Implemented as pypgx run-chip-pipeline in CLI and
pypgx.pipeline.run_chip_pipeline in API, this pipeline is designed for
DNA chip data (e.g. Global Screening Array from Illumina). It’s recommended
to perform variant imputation on the input VCF prior to feeding it to the
pipeline using a large reference haplotype panel (e.g. TOPMed Imputation
Server).
Alternatively, it’s possible to perform variant imputation with the 1000
Genomes Project (1KGP) data as reference within PyPGx using --impute in
CLI and impute=True in API.
The pipeline currently does not support SV detection. Please post a GitHub issue if you want to contribute your development skills and/or data for devising an SV detection algorithm.
Check out the Coriell Affy tutorial to see this pipeline in action.
Long-read pipeline
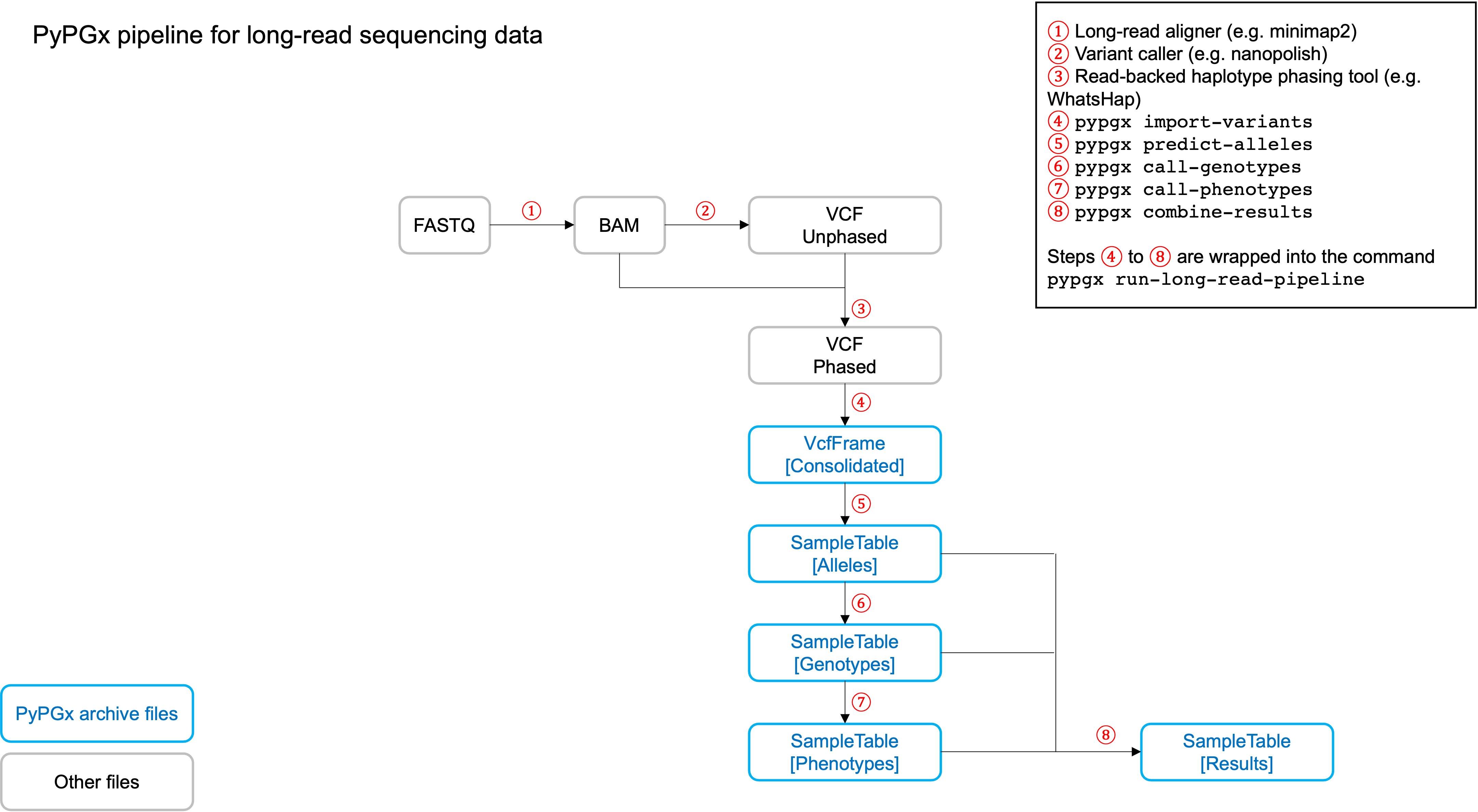
Implemented as pypgx run-long-read-pipeline in CLI and
pypgx.pipeline.run_long_read_pipeline in API, this pipeline is designed
for long-read data (e.g. Pacific Biosciences and Oxford Nanopore
Technologies). The input VCF must be phased using a read-backed haplotype
phasing tool such as WhatsHap.
The pipeline currently does not support SV detection. Please post a GitHub issue if you want to contribute your development skills and/or data for devising an SV detection algorithm.
Results interpretation
PyPGx outputs per-sample genotype results in a table, which is stored in an
archive file with the semantic type SampleTable[Results]. Below, we will
use the CYP2D6 gene with GRCh37 as an example to illustrate how to interpret
genotype results from PyPGx.
Genotype |
Phenotype |
Haplotype1 |
Haplotype2 |
AlternativePhase |
VariantData |
CNV |
|
|---|---|---|---|---|---|---|---|
NA11839 |
*1/*2 |
Normal Metabolizer |
*1; |
*2; |
; |
*1:22-42522613-G-C,22-42523943-A-G:0.5,0.488;*2:default |
Normal |
NA12006 |
*4/*41 |
Intermediate Metabolizer |
*41;*2; |
*4;*10;*2; |
*69; |
*69:22-42526694-G-A,22-42523805-C-T:0.5,0.551;*4:22-42524947-C-T:0.444;*10:22-42523943-A-G,22-42526694-G-A:0.55,0.5;*41:22-42523805-C-T:0.551;*2:default; |
Normal |
HG00276 |
*4/*5 |
Poor Metabolizer |
*4;*10;*74;*2; |
*10;*74;*2; |
; |
*4:22-42524947-C-T:0.913;*10:22-42523943-A-G,22-42526694-G-A:1.0,1.0;*74:22-42525821-G-T:1.0;*2:default; |
WholeDel1 |
NA19207 |
*2x2/*10 |
Normal Metabolizer |
*10;*2; |
*2; |
; |
*10:22-42523943-A-G,22-42526694-G-A:0.361,0.25;*2:default; |
WholeDup1 |
This list explains each of the columns in the example results.
Genotype: Diplotype call. When there is no SV this simply combines the two top-ranked star alleles from Haplotype1 and Haplotype2 with the delimiter ‘/’. In the presence of SV the final diplotype is determined using one of the genotypers in the
pypgx.api.genotypemodule (e.g. CYP2D6Genotyper).Phenotype: Phenotype call.
Haplotype1, Haplotype2: List of candidate star alleles for each haplotype. For example, if a given haplotype contains three variants
22-42523943-A-G,22-42524947-C-T, and22-42526694-G-A, then it will get assigned*4;*10;because the haplotype pattern can fit both *4 (22-42524947-C-T) and *10 (22-42523943-A-Gand22-42526694-G-A). Note that *4 comes first before *10 because it has higher priority for reporting purposes (see thepypgx.sort_allelesmethod for detailed implementation).AlternativePhase: List of star alleles that could be missed due to potentially incorrect statistical phasing. For example, let’s assume that statistical phasing has put
22-42526694-G-Afor Haplotype1 and22-42523805-C-Tfor Haplotype2. Even though the two variants are in trans orientation, PyPGx will also consider alternative phase in case the two variants are actually in cis orientation, resulting in*69;as AlternativePhase because *69 is defined by22-42526694-G-Aand22-42523805-C-T.VariantData: Information for SNVs/indels used to define observed star alleles, including allele fraction which is important for allelic decomposition after identifying CNV (e.g. the sample NA19207). In some situations, there will not be any variants for a given star allele because the allele itself is “default” allele for the selected reference assembly (e.g. GRCh37 has *2 as default while GRCh38 has *1).
CNV: Structural variation call. See the Structural variation detection section for more details.
Getting help
For detailed documentations on the CLI and API, please refer to the Read the Docs.
For getting help on the CLI:
$ pypgx -h
usage: pypgx [-h] [-v] COMMAND ...
positional arguments:
COMMAND
call-genotypes Call genotypes for target gene.
call-phenotypes Call phenotypes for target gene.
combine-results Combine various results for target gene.
compare-genotypes Calculate concordance between two genotype results.
compute-control-statistics
Compute summary statistics for control gene from BAM
files.
compute-copy-number
Compute copy number from read depth for target gene.
compute-target-depth
Compute read depth for target gene from BAM files.
create-consolidated-vcf
Create a consolidated VCF file.
create-input-vcf Call SNVs/indels from BAM files for all target genes.
create-regions-bed Create a BED file which contains all regions used by
PyPGx.
estimate-phase-beagle
Estimate haplotype phase of observed variants with
the Beagle program.
filter-samples Filter Archive file for specified samples.
import-read-depth Import read depth data for target gene.
import-variants Import SNV/indel data for target gene.
plot-bam-copy-number
Plot copy number profile from CovFrame[CopyNumber].
plot-bam-read-depth
Plot read depth profile with BAM data.
plot-cn-af Plot both copy number profile and allele fraction
profile in one figure.
plot-vcf-allele-fraction
Plot allele fraction profile with VCF data.
plot-vcf-read-depth
Plot read depth profile with VCF data.
predict-alleles Predict candidate star alleles based on observed
variants.
predict-cnv Predict CNV from copy number data for target gene.
prepare-depth-of-coverage
Prepare a depth of coverage file for all target
genes with SV from BAM files.
print-data Print the main data of specified archive.
print-metadata Print the metadata of specified archive.
run-chip-pipeline Run genotyping pipeline for chip data.
run-long-read-pipeline
Run genotyping pipeline for long-read sequencing data.
run-ngs-pipeline Run genotyping pipeline for NGS data.
slice-bam Slice BAM file for all genes used by PyPGx.
test-cnv-caller Test CNV caller for target gene.
train-cnv-caller Train CNV caller for target gene.
options:
-h, --help Show this help message and exit.
-v, --version Show the version number and exit.
For getting help on a specific command (e.g. call-genotypes):
$ pypgx call-genotypes -h
Below is the list of submodules available in the API:
core : The core submodule is the main suite of tools for PGx research.
genotype : The genotype submodule is primarily used to make final diplotype calls by interpreting candidate star alleles and/or detected structural variants.
pipeline : The pipeline submodule is used to provide convenient methods that combine multiple PyPGx actions and automatically handle semantic types.
plot : The plot submodule is used to plot various kinds of profiles such as read depth, copy number, and allele fraction.
utils : The utils submodule contains main actions of PyPGx.
For getting help on a specific submodule (e.g. utils):
>>> from pypgx.api import utils
>>> help(utils)
For getting help on a specific method (e.g. pypgx.predict_phenotype):
>>> import pypgx
>>> help(pypgx.predict_phenotype)
In Jupyter Notebook and Lab, you can see the documentation for a python
function by hitting SHIFT + TAB. Hit it twice to expand the view.
CLI examples
We can print the metadata of an archive file:
$ pypgx print-metadata grch37-depth-of-coverage.zip
Above will print:
Assembly=GRCh37
SemanticType=CovFrame[DepthOfCoverage]
Platform=WGS
We can run the NGS pipeline for the CYP2D6 gene:
$ pypgx run-ngs-pipeline \
CYP2D6 \
grch37-CYP2D6-pipeline \
--variants grch37-variants.vcf.gz \
--depth-of-coverage grch37-depth-of-coverage.zip \
--control-statistics grch37-control-statistics-VDR.zip
Above will create a number of archive files:
Saved VcfFrame[Imported] to: grch37-CYP2D6-pipeline/imported-variants.zip
Saved VcfFrame[Phased] to: grch37-CYP2D6-pipeline/phased-variants.zip
Saved VcfFrame[Consolidated] to: grch37-CYP2D6-pipeline/consolidated-variants.zip
Saved SampleTable[Alleles] to: grch37-CYP2D6-pipeline/alleles.zip
Saved CovFrame[ReadDepth] to: grch37-CYP2D6-pipeline/read-depth.zip
Saved CovFrame[CopyNumber] to: grch37-CYP2D6-pipeline/copy-number.zip
Saved SampleTable[CNVCalls] to: grch37-CYP2D6-pipeline/cnv-calls.zip
Saved SampleTable[Genotypes] to: grch37-CYP2D6-pipeline/genotypes.zip
Saved SampleTable[Phenotypes] to: grch37-CYP2D6-pipeline/phenotypes.zip
Saved SampleTable[Results] to: grch37-CYP2D6-pipeline/results.zip
API examples
We can obtain allele function for the CYP2D6 gene:
>>> import pypgx
>>> pypgx.get_function('CYP2D6', '*1')
'Normal Function'
>>> pypgx.get_function('CYP2D6', '*4')
'No Function'
>>> pypgx.get_function('CYP2D6', '*22')
'Uncertain Function'
>>> pypgx.get_function('CYP2D6', '*140')
'Unknown Function'
We can predict phenotype for CYP2D6 based on two haplotype calls:
>>> import pypgx
>>> pypgx.predict_phenotype('CYP2D6', '*4', '*5') # Both alleles have no function
'Poor Metabolizer'
>>> pypgx.predict_phenotype('CYP2D6', '*5', '*4') # The order of alleles does not matter
'Poor Metabolizer'
>>> pypgx.predict_phenotype('CYP2D6', '*1', '*22') # *22 has uncertain function
'Indeterminate'
>>> pypgx.predict_phenotype('CYP2D6', '*1', '*1x2') # Gene duplication
'Ultrarapid Metabolizer'
We can also obtain recommendation (e.g. CPIC) for certain drug-phenotype combination:
>>> import pypgx
>>> # Codeine, an opiate and prodrug of morphine, is metabolized by CYP2D6
>>> pypgx.get_recommendation('codeine', 'CYP2D6', 'Normal Metabolizer')
'Use codeine label recommended age- or weight-specific dosing.'
>>> pypgx.get_recommendation('codeine', 'CYP2D6', 'Ultrarapid Metabolizer')
'Avoid codeine use because of potential for serious toxicity. If opioid use is warranted, consider a non-tramadol opioid.'
>>> pypgx.get_recommendation('codeine', 'CYP2D6', 'Poor Metabolizer')
'Avoid codeine use because of possibility of diminished analgesia. If opioid use is warranted, consider a non-tramadol opioid.'
>>> pypgx.get_recommendation('codeine', 'CYP2D6', 'Indeterminate')
'None'